Condensation and localization of the partitioning protein ParB on the bacterial chromosome
- Chase P. Broedersza,b,
- Xindan Wangc,
- Yigal Meird,
- Joseph J. Loparoe,
- David Z. Rudnerc, and
- Ned S. Wingreena,f,1
-
Edited by Ken A. Dill, Stony Brook University, Stony Brook, NY, and approved May 1, 2014 (received for review February 10, 2014)
Significance
The ParABS system is responsible for chromosome and plasmid segregation in many bacteria. A large, coherent ParB–DNA complex forms the partitioning module at the heart of this segregation machinery. Here we provide a simple theoretical model for interacting proteins on DNA to elucidate the structure of the ParB–DNA complex. We show that that both 3D bridging and 1D spreading interactions between DNA-bound ParB proteins are required to ensure the formation of a coherent protein–DNA complex. This combination of protein–protein interactions implies a surface tension that drives the condensation of ParB proteins on the DNA. The formation of such a condensed protein complex is essential for understanding how a single centromeric parS site can localize ParB on the DNA.
Abstract
The ParABS system mediates chromosome segregation and plasmid partitioning in many bacteria. As part of the partitioning mechanism, ParB proteins form a nucleoprotein complex at parS sites. The biophysical basis underlying ParB–DNA complex formation and localization remains elusive. Specifically, it is unclear whether ParB spreads in 1D along DNA or assembles into a 3D protein–DNA complex. We show that a combination of 1D spreading bonds and a single 3D bridging bond between ParB proteins constitutes a minimal model for a condensed ParB–DNA complex. This model implies a scaling behavior for ParB-mediated silencing of parS-flanking genes, which we confirm to be satisfied by experimental data from P1 plasmids. Furthermore, this model is consistent with experiments on the effects of DNA roadblocks on ParB localization. Finally, we show experimentally that a single parS site is necessary and sufficient for ParB–DNA complex formation in vivo. Together with our model, this suggests that ParB binding to parS triggers a conformational switch in ParB that overcomes a nucleation barrier. Conceptually, the combination of spreading and bridging bonds in our model provides a surface tension ensuring the condensation of the ParB–DNA complex, with analogies to liquid-like compartments such as nucleoli in eukaryotes.
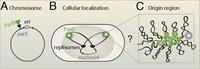
Chromosomal organization and segregation presents a major challenge in all organisms. Partitioning proteins of the ParABS system play a key role in chromosomal segregation and mediate plasmid partitioning in a variety of bacteria, including Caulobacter crescentus, Bacillus subtilis, and Vibrio cholerae (1, 2). This partitioning module includes a DNA binding protein (ParB) that forms a large nucleoprotein complex at centromere-like parS sites, frequently located near the origin of replication (Fig. 1) (3). These ParB–DNA complexes interact with ParA ATPases, leading to segregation of replicated origins (4⇓⇓⇓–8). Despite the apparent simplicity of this segregation machinery, puzzles remain: What is the nature of interactions among DNA-bound ParB proteins, and how do these determine the organizational and functional properties of the ParB–DNA partitioning complex? A central question is whether ParB spreads along the DNA to form a 1D filamentous protein–DNA complex or assembles into a 3D complex on the DNA. Furthermore, it remains unclear how a small number of parS sites (typically 2–10) leads to robust formation and localization of such a large protein–DNA complex.
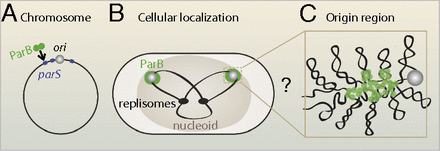
Cellular localization of ParB. (A) ParB binding sites (parS) are frequently present near the replication origin (ori). (B) In cells, GFP-ParB proteins form fluorescent foci that colocalize with replication origins. The mass of DNA (the nucleoid) is shown schematically as a large oval, including a simplified view of the replicating chromosome (black lines) and the replisomes (black ovals). (C) A hypothetical magnified view of the origin region where ParB forms a large protein–DNA complex.
Live cell microscopy experiments indicate that ParB-GFP fusion proteins form a large fluorescent focus on the bacterial chromosome (6, 7, 9⇓⇓–12). ParB–DNA complexes do not rely on the presence of ParA or on the particular DNA sequence other than the parS site (12⇓–14). Genome-wide chromatin immunoprecipitation studies (ChIP-chip) in B. subtilis have shown that ParB (Spo0J) binds site-specifically to eight origin-proximal parS sites (13) and also revealed significantly enhanced binding to DNA in the vicinity of each parS site up to distances of 18 kbp. The association of ParB proteins with sites surrounding parS is often referred to as spreading. However, for the sake of clarity, here we refer to this as the formation of ParB–DNA complexes. We reserve the word “spreading” to describe a purely 1D coating of ParB along DNA to form a nucleoprotein filament. The actual structure of the ParB–DNA complexes remains unclear.
Experiments on P1 plasmids have shown that ParB overexpression leads to silencing of parS-proximal genes (15). However, in many cases gene silencing is only partial and strongly dependent on genomic distance from a parS site, suggesting that the ParB–DNA complex is partially accessible to the transcriptional machinery. Indeed, transcription of genes adjacent to parS sites in B. subtilis is virtually unaffected by Spo0J complex formation at native Spo0J expression levels (13).
The formation of a ParB–DNA complex at parS suggests the presence of ParB–ParB interactions. In vitro experiments indicate that ParB is largely dimerized in solution owing to its C-terminal dimerization domain. Moreover, the ParB crystal structure from Thermus thermophilus suggests an N-terminal interface that acts as a secondary dimerization domain between DNA-bound ParB proteins (16). In support of this idea, a Spo0J93 mutant that contains a single amino acid substitution near the N terminus (G77S), has wild-type nonspecific and specific DNA binding affinities in vitro but lacks the ability to form ParB–DNA complexes surrounding parS sites on the chromosome in vivo and fails to form discrete fluorescent foci (13). Taken together with the structural data, this suggests that in addition to a C-terminal dimerization domain, ParB (Spo0J) also has an N-terminal interaction domain, which is required to form a ParB–DNA complex.
Further insight into the nature of ParB–ParB interactions was gleaned from roadblock experiments (15, 17) in which a strong binding site for a transcriptional repressor was inserted near a parS site. In B. subtilis the bound repressor leads to a reduction in ParB binding to DNA adjacent to parS, but only in the direction of the roadblock (17). This was taken as evidence that ParB associates with the DNA by 1D lateral spreading from parS to form a nucleoprotein filament. Alternatively, ParB may form higher-order interactions resulting in a 3D protein–DNA complex.
To investigate the structure of the ParB partitioning complex, we developed a simple model for interacting proteins on DNA. We found that a combination of 1D spreading bonds and a 3D bridging bond between ParB proteins constitutes the minimal model for condensation of ParB proteins on DNA into a coherent complex. These combined interactions provide an effective surface tension, preventing fragmentation of the complex. In detail, our model predicts that ParB spreads to form multiple, short 1D domains on the DNA, connected in 3D by bridging interactions to assemble into a 3D ParB–DNA condensate (Fig. 1C). More generally, the computational model we developed here offers a simple framework to study how various interactions between DNA-binding proteins determine the structure and localization of protein–DNA complexes.
Model for Interacting Proteins on DNA
We developed a minimal model to investigate the spatial organization of interacting proteins on DNA. For simplicity, the DNA
is described as a linear, self-avoiding chain on a cubic lattice in 3D; the DNA chain has a bending stiffness κ and N protein binding sites (Supporting Information). The addition of DNA confinement has little effect on our central results (Supporting Information). The DNA is coarse-grained at the scale of a protein-binding site,
Proteins can bind or unbind the DNA and the binding energy may vary along the DNA. Here, we considered a chain with equivalent
binding sites, or a chain with just one cognate parS site with a binding energy
Results
For many bacteria, the faithful partitioning and segregation of plasmids and the chromosome relies on ParB proteins, which form large ParB–DNA complexes localized by a few or in rare cases by one parS site—a 16-bp sequence that specifically binds a ParB dimer (3). We first discuss how various types of protein–protein interactions in our model would affect the structure and stability of the ParB–DNA complex in the absence of a parS site. We then proceed to include a parS site in our model to investigate how this affects the localization of the ParB cluster on the DNA, and finally we consider the role of the parS site in ParB cluster nucleation.
Combining 1D Spreading with 3D Bridging Interactions Is Necessary for the Formation of a Condensed Protein–DNA Complex.
To serve as a benchmark, we first defined a dimer model, in which DNA-bound proteins interact through their primary dimerization domain (Fig. 2A). An additional dimerization domain on the protein (16, 18) could, in principle, engage in either 1D spreading or 3D bridging interactions leading to two additional models: a spreading model (Fig. 2B) in which each protein in the ParB dimer can form two 1D spreading interactions, one in each direction along the chain, and a bridging model (Fig. 2C), in which each dimer can engage in a single 3D bridging interaction.
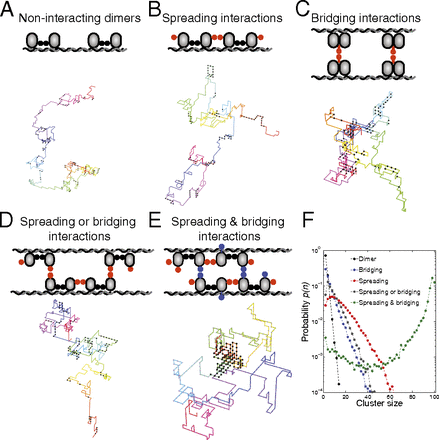
Simple models for interacting DNA-bound proteins. Schematics include a representative image of a DNA–protein complex from
simulations: (A) dimer model, (B) spreading model, (C) bridging model, (D) spreading or bridging model, and (E) spreading and bridging model. In the spreading or bridging model there are two interaction domains per monomer, one of which
can be either in spreading or in bridging mode. In the spreading and bridging model there are two 1D spreading bonds along
the DNA and one 3D bridging bond. The bond energies are
The dimer, spreading, and bridging models all resulted in multiple protein clusters of varying sizes dispersed over the DNA
(with no parS site), as shown in representative images of simulated protein–DNA complexes in Fig. 2 A–C. To quantify the distribution of cluster sizes in a system with a fixed number of proteins, we evaluated the probability
Conceptually, the fragmentation in these models can be traced to the entropy of dispersing the proteins over the DNA, which is favored over the energy of forming a single large cluster. To account for the experimental observation of coherent ParB–DNA foci (6, 7, 9⇓⇓–12), we expanded the model by combining 1D spreading and 3D bridging interactions to avoid entropic fragmentation. In the simplest of such models, the proteins still only have two interaction domains, as in the spreading model, but now one of the interaction domains can either function in spreading or in bridging mode, whereas the other domain can only participate in a 1D spreading bond (Fig. 2D). Importantly, this model failed to prevent fragmentation because the distribution of cluster sizes was broad and qualitatively similar to the previous models (Fig. 2F).
We next considered a model with one additional ingredient: each ParB protein is able to interact with its two neighbors via 1D spreading interactions along with an additional 3D bridging interaction. Strikingly, in this spreading and bridging model the majority of the proteins cluster together in one coherent focus on the DNA (Fig. 2E). This is reflected as a large, narrow peak centered around the maximal number of proteins in the cluster size distribution (Fig. 2F).
Importantly, for the spreading and bridging model, unsatisfied dangling ParB bonds at the surface of the cluster generate a surface tension that counters the tendency of entropy to fragment the condensate into multiple small clusters. By contrast, the spreading model cannot support a condensed phase because 1D surface tension always loses to entropy. Moreover, for the bridging model and spreading or bridging model the most stable state consists of small clusters with no surface tension because all bonds can be satisfied. (A variety of other models can be excluded for similar reasons) (Supporting Information). Thus, the combination of both spreading and bridging interactions constitutes the minimal requirement for a condensed phase (i.e., a large, coherent ParB–DNA cluster). These conclusions also hold when we include DNA confinement in our model (Supporting Information).
A parS Site Can Localize a Condensed ParB Complex.
DNA-bound ParB proteins localize around parS sites. ParB binds specifically to a parS site, but in vitro experiments suggest that the binding affinity to a parS site is only roughly 10-fold higher than to nonspecific DNA (13). Thus, it is unclear how a few parS sites can localize the majority of ParB proteins.
To investigate the effects of a parS site on the organization of ParB on the DNA, we inserted a site at the center of the DNA in our model with a strong binding
energy
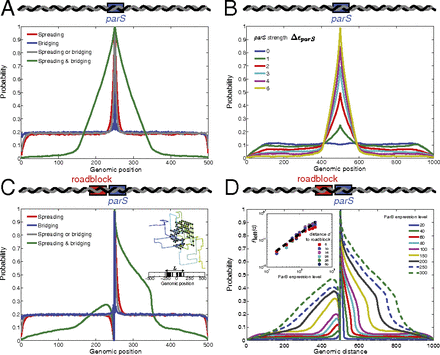
ParB binding profiles, ParB localization, and the effects of a roadblock. (A) Probability of a bound protein versus genomic position with a central parS site (parameters as in Fig. 2 with
Roadblocks Strongly Affect the Localization of 3D ParB–DNA Complexes.
In B. subtilis, the insertion of a strong binding site for a transcriptional repressor in the vicinity of a parS site led to a reduction in ParB–DNA interaction in the direction of the repressor bound site (17). Based on this observation, it was proposed that ParB associates with the DNA by spreading one-dimensionally from a parS site. To investigate the effect of a roadblock on the binding profiles in our models, we inserted a blocked site, to which ParB is not allowed to bind, immediately to the left of the parS site (Fig. 3C). The binding profile of the bridging model is hardly affected by the roadblock, whereas for the spreading model and the spreading or bridging model the binding of ParB is obstructed by the roadblock. Strikingly, the binding profile is also strongly asymmetric for the spreading and bridging model, consistent with the observations from roadblock experiments in B. subtilis (17).
The small binding probability of ParB to the left of the roadblock for the spreading and bridging model indicates that there
are some configurations in which the ParB cluster forms loops that bypass the roadblock. This raises the question why it is
thermodynamically more favorable for the complex to stay to the parS side of the roadblock instead of looping around it. To investigate this, we analyzed the loops extruding from the surface
of the cluster (Fig. 3C, Inset). Interestingly, we find that the average loop length
Intuitively, the greater number of loops in a larger cluster together with the greater number of configurations a larger cluster
can adopt on the DNA should render it less sensitive to the presence of a roadblock. Indeed, our simulations show that the
binding profiles become more symmetric with increasing ParB expression levels (Fig. 3D), indicating that it becomes more likely to loop around the roadblock for larger clusters. Fig. 3D, Inset demonstrates that the ParB binding probability to the left of the roadblock scales as
Scaling Behavior for ParB-Induced Gene Silencing.
Experiments on P1 plasmids indicate that ParB binding to DNA can partially silence genes depending on their distance from parS and on ParB expression levels (15). In contrast, it was found in B. subtilis that transcription was unaffected by the formation of Spo0J nucleoprotein complexes at normal Spo0J expression levels (13). These experiments raise the question of how the presence of a 3D ParB–DNA complex affects the transcriptional machinery.
To model the effects of a ParB–DNA complex on transcription, we first obtained average parS-proximal binding profiles of ParB on the DNA as a function of ParB expression levels using the spreading and bridging model. Fig. 4A shows how the binding profile broadens with increasing ParB levels, while the shape remains roughly constant. This suggests a rescaling of the genomic distance from parS by the ParB expression level; such a rescaling results in a data collapse, as shown in Fig. 4B (Supporting Information).

The effects of ParB cluster formation on gene silencing. (A) Probability of a bound ParB versus genomic position for the spreading and bridging model with a central parS site (
What does this scaling property of the ParB profile mean for the effects of ParB–DNA complex formation on gene silencing?
We used the exposure of the DNA (defined as 1 − ParB binding probability) as a simple proxy for the remaining activity of
a gene at a particular distance from the parS site, defined relative to the activity of a gene without ParB. To compare with the experiments of Rodionov et al. (15) using plasmid P1, we plotted the DNA exposure as a function of ParB expression levels for varying genomic distances from
the parS site, as shown in Fig. 4C. These simulation results can also be collapsed for a fixed value of the spreading bond strength
The Role of the parS Site in ParB–DNA Complex Formation and Localization.
Interestingly, ParB does not condense into foci in cells lacking parS sites (Fig. 5C and Supporting Information and ref. 12). This is perplexing in light of in vitro experiments showing that the binding affinity of ParB to a parS site is only 10-fold higher than to nonspecific DNA (13).
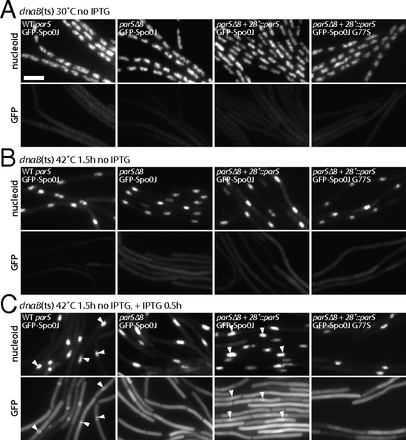
A single parS site is necessary and sufficient to generate a GFP-Spo0J focus. (A) Representative images of B. subtilis cells harboring a dnaB(ts) allele with all eight wild-type parS sites (BWX2454, WT parS, first panel); none of the eight endogenous parS sites (BWX2456, parSΔ8, second panel); parSΔ8 with an ectopic parS site inserted at 28° (amyE) (BWX2458 and BWX2789, parSΔ8+28°::parS, third and fourth panels) grown at 30 °C (A) and after growth at 42 °C for 1.5 h (B) to block new rounds of initiation of replication generating a single chromosome (Supporting Information). Expression of GFP-Spo0J (first to third panels) or GFP-Spo0J (G77S) (fourth panel) was then induced by the addition of IPTG (0.5 mM final concentration) for 0.5 h at 42 °C (C) (see Supporting Information for induction control). GFP-Spo0J did not form foci in the strain lacking parS sites (parSΔ8, second panel) and formed a single focus per nucleoid in the parSΔ8+28°::parS strain (third panel). GFP-Spo0J (G77S) did not form foci in theparSΔ8+28°::parS strain (fourth panel). Based on comparison with Spo0J (G77S) (C, fourth panel) that binds parS but is unable to spread to neighboring sites (13), the foci observed in the parSΔ8+28°::parS strain (C, third panel) reflect GFP-Spo0J nucleoprotein complexes. Gray scale of DAPI-stained nucleoids (upper panels) and GFP (lower panels) is shown. GFP-Spo0J foci (white carets) are highlighted in the DAPI and GFP panels. (Scale bar, 4 μm.)
We can rule out a purely thermodynamic basis for the mechanism by which a parS site robustly produces a ParB–DNA complex. We found that, in general, the formation of a ParB–DNA complex in the spreading
and bridging model does not rely on the presence of a parS site (Fig. 2 E and F). The affinity of a parS site contributes at most several
Nucleation kinetics offers an alternative, robust mechanism to account for the role of a parS site in cluster formation. Even if cells always operate under conditions thermodynamically favoring the ParB–DNA condensed state, substantial nucleation barriers can kinetically impede the formation of a ParB–DNA condensate. We can envision two scenarios for how ParB/parS functions to overcome a nucleation barrier. In the first, two (or more) ParB dimers bound to nearby parS sites form a nucleation center from which ParB–DNA complexes grow (Supporting Information). The relative ParB binding affinity to a parS site is far too low for this kind of nucleation to occur reliably at a single parS site but not elsewhere on the DNA, as explained further below. In the second scenario, nucleation occurs at a single parS site because some characteristic of ParB when bound to a parS site dramatically enhances the rate of complex nucleation.
To distinguish between these two models, we sought to determine whether a ParB–DNA complex (or a GFP-ParB focus) could be generated in cells that contain only one parS site. Indeed, previous cytological experiments indicate that B. subtilis cells that harbor a single parS site form GFP-Spo0J fluorescent foci (12). However, the cells analyzed in these experiments were actively growing and therefore undergoing DNA replication. Accordingly, before their segregation, the replicated sister parS sites could together serve as a nucleation center for ParB–DNA complexes. To more rigorously test whether a single parS site was sufficient to generate a ParB–DNA complex, we constructed a set of strains harboring a temperature-sensitive replication initiation mutant (dnaBts) (20) to block new rounds of DNA replication. These strains also contained an isopropyl β-d-1-thiogalactopyranoside (IPTG)-inducible GFP-Spo0J fusion as the sole source of Spo0J (ParB), allowing conditional expression of GFP-Spo0J after replication had ceased. We compared a strain that had the full complement of endogenous parS sites (WT parS), a strain lacking all eight parS sites (parSΔ8), and one in which a single ectopic parS site was inserted at a nonessential locus (parSΔ8+28°::parS). Finally, we also constructed a matched control strain harboring a single parS site, containing an IPTG-inducible GFP-spo0J mutant (G77S) that binds parS in vivo but does not form a ParB–DNA complex (14). When grown at the permissive temperature (30 °C) in the absence of IPTG, all four strains contained DAPI-stained chromosomes (called nucleoids) that resembled the chromosomes observed in the Spo0J null mutant (Fig. 5A). Importantly, GFP-Spo0J fluorescence was virtually undetectable (Fig. 5A). After 1.5 h of growth at the restrictive temperature (42 °C), virtually all cells in the four strains contained a single replicated chromosome (Fig. 5B and Supporting Information). At this time, IPTG was added to induce expression of GFP-Spo0J. Thirty minutes later, GFP-Spo0J localization was monitored. As expected, the cells harboring the endogenous origin-proximal parS sites had a single fluorescent focus (or cluster of foci) that colocalized with the nucleoid (Fig. 5C). Moreover, the cells lacking parS sites had no detectable GFP-Spo0J foci. By contrast, virtually all cells with a single parS site had a faint GFP-Spo0J focus that colocalized with the nucleoid. These foci correspond to Spo0J-DNA condensates and not simply a Spo0J dimer bound to the parS site because no foci were detected in the GFP-Spo0J (G77S) mutant that binds parS but does not form a nucleoprotein complex (Fig. 5C). Immunoblot analysis indicates that the GFP-Spo0J fusions were expressed at similar levels in the different strains and the cytoplasmic fluorescence was not due to proteolytic release of GFP (Supporting Information). Taken together, these data indicate that a single parS site per cell is capable of forming a Spo0J-DNA complex, whereas no complex forms in the absence of a parS site. We therefore favor a model in which a Spo0J dimer bound to a parS site is able to nucleate a Spo0J–DNA complex much more rapidly than a Spo0J dimer bound elsewhere.
Importantly, the requisite parS-induced reduction of the nucleation free-energy barrier,
However, the relative affinity of specific binding of ParB (Spo0J) to parS binding was found to be only 10-fold higher than nonspecific binding (13), corresponding to
Discussion
In summary, we have identified a minimal model for interacting ParB proteins on DNA that produces large, coherent ParB–DNA complexes, as observed in live cells (6, 7, 9⇓⇓–12). This model offers a conceptual framework in which a combination of 1D spreading and 3D bridging protein–protein interactions thermodynamically stabilizes a ParB–DNA condensate. Put simply, we find that both spreading and bridging interactions are required to provide a surface tension that prevents fragmentation of condensed protein–DNA complexes. Indeed, we speculate that the ParB–DNA complex may organize like a liquid-like (non–membrane-bound) compartment analogous to, e.g., the nucleolus in eukaryotes (22, 23).
The spreading and bridging model accounts for multiple experimental observations, including ParB reorganization by DNA roadblocks
and ParB-induced gene silencing. What aspects of the model are essential to explain these experiments? The roadblock experiments
indicate the presence of strong spreading bonds between ParB proteins (17), and the formation and localization of a coherent ParB–DNA complex (6, 7, 9⇓⇓–12) requires an additional bridging bond between proteins. Crystallographic studies of ParB proteins indicate that there are
at least two interaction domains: one at the C terminus that likely promotes dimerization and one at the N terminus (16). An N-terminal Spo0J mutant (G77S) binds parS in vivo but fails to form nucleoprotein complexes. Accordingly, this domain is likely to function in either 1D spreading
or 3D bridging. Our model predicts that each ParB monomer has three interaction domains, and thus can form two spreading interactions
along the DNA as well as one bridging interaction with another DNA-bound ParB protein, resulting in the formation of clusters
consisting of multiple 1D spreading domains of ParB connected in 3D by bridging interactions to form a coherent ParB–DNA complex.
These structural predictions could be tested by single-molecule pulling experiments on condensed ParB–DNA complexes. Recent
single-molecule experiments using flow-stretched DNA provide evidence for bridging interactions between ParB proteins in vitro,
consistent with our model (24). These experiments also show that ParB is capable of condensing DNA with rates up to
We showed experimentally that a single parS site is both necessary and sufficient for nucleation of a ParB–DNA complex. These experiments together with calculations
based on our model led us to conclude that binding of ParB to a parS site effectively lowers the nucleation barrier, leading reliably to complex nucleation at parS sites but not at nonspecific sites. Curiously, we find that the requisite parS-induced reduction of the nucleation barrier has to be much larger than the energy difference inferred from the measured binding
affinity of ParB to parS versus nonspecific sites. What mechanism could give rise to a dramatically reduced nucleation barrier at a parS site? It has been speculated based on structural data that the ability of DNA-bound ParB proteins to interact may require
a conformational change (16). This proposed conformational transition may involve breaking the primary C-terminal dimerization bond. Thus, there may
be two binding modes: ParB could bind to the DNA as closed inert dimers as in our dimer model, or as open dimers as in our
spreading and bridging model (Fig. 2 A and E). Building on this idea, we conjecture that the transition to the open configuration is favored at a parS site, whereas the closed configuration is favored at nonspecific sites. An estimate based on this scenario is that the nucleation
barrier at a parS site, relative to nonspecific sites, is lowered by
The biophysical properties of the ParB–DNA complex implied by our model may have important implications for the segregation and organization of the chromosome. The surface tension of the ParB–DNA complex in the spreading and bridging model indicates that it is thermodynamically most favorable to form a single ParB–DNA complex that recruits all ori-proximal parS sites. Our model highlights a puzzling aspect of the observed splitting of the parB–DNA complex during segregation (6, 7, 9⇓⇓–12), which would be disfavored by the surface tension of the ParB–DNA complex. This suggests that the splitting of the ParB–DNA complex may rely on active processes, in the case of B. subtilis, likely mediated by the ParA ATPase and the action of structural maintenance of chromosomes condensin complexes (26, 27).
Our model may also help provide insights into the organization of DNA by nucleoid-associated proteins (NAPs). The model offers a simple framework to study how the specific types of protein-protein interactions in NAPs determine the structure and function of protein–DNA complexes.
Materials and Methods
Bacteria Strains and Growth.
All B. subtilis strains were derived from the prototrophic strain PY79. Cells were grown in defined rich casein hydrolysate medium (28) at indicated temperatures. Strains, plasmids, oligonucleotide primers, and our data analysis are described in Supporting Information.
Acknowledgments
We thank B. Bratton, Z. Gitai, B. Machta, and M. Tikhonov for insightful discussions. This work was supported in part by a Lewis–Sigler fellowship (to C.P.B.), National Science Foundation Grant PHY-0957573 (to N.S.W.), National Institutes of Health Grants GM086466 and GM073831 (to D.Z.R.), National Science Foundation CAREER Award MCB-1148818 (to J.J.L.), the Human Frontier Science Program (X.W.), and the hospitality of the Aspen Center for Physics (National Science Foundation Grant PHYS-1066293).
Footnotes
- ↵1To whom correspondence should be addressed. E-mail: wingreen@princeton.edu.
-
Author contributions: C.P.B., J.J.L., D.Z.R., and N.S.W. designed research; C.P.B. and X.W. performed research; C.P.B., X.W., Y.M., J.J.L., D.Z.R., and N.S.W. analyzed data; and C.P.B., D.Z.R., and N.S.W. wrote the paper.
-
The authors declare no conflict of interest.
-
This article is a PNAS Direct Submission.
-
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1402529111/-/DCSupplemental.